
Forschungsbericht
Epidemiologie
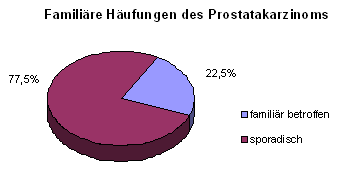
Wir stehen mit 279 urologischen Kliniken, 2263 niedergelassenen Urologen und 5 Tumorzentren in Kontakt. In unserer Datenbank sind derzeit 28890 Patienten aus 22345 Familien aufgenommen. In 17307 (77,5%) Familien liegt die Erkrankung als sporadische Form vor. 5038 (22,5%) Familien gaben eine familiäre Disposition (mindestens 1 zusätzlicher Angehöriger mit PCA) an.
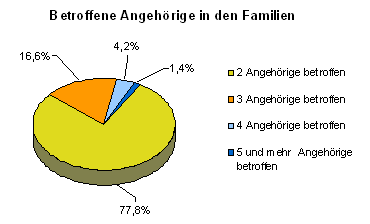
Dabei waren in 3920 (77,8%) Familien 2 Angehörige, in 836 (16,6%) Familien 3 Angehörige, in 213 (4,2%) Familien 4 Angehörige und in 49 (0,97%) Familien 5 Angehörige betroffen. 12 Familien (0,24%) wiesen 6 und 4 (0,08%) Familien 7 Fälle mit Prostatakarzinom auf. In jeweils 2 (0,04%) Familien waren 8 bzw. 11 Angehörige betroffen. Die Kriterien für das hereditäre Prostatakarzinom nach Carter (3 Angehörige I. Grades oder 3 Angehörige in 3 verschiedenen Generationen oder 2 Angehörige I. Grades mit Erkrankungsalter < 55) waren bei 6,2% der betroffenen Familien erfüllt.
Genetik
Assoziationsstudien Kopplungsanalysen Funktionelle Analysen
Allgemeines
Im wesentlichen gibt es zwei Untersuchungsansätze, um in einer Situation wie beim Prostata-Karzinom (eine Vielzahl sporadischer Fälle steht einem Anteil genetisch bedingter familiärer gegenüber) genetische Faktoren zu identifizieren, wobei beide Ansätze auf unterschiedliche Untersuchungmaterialien (Probandenkollektive) zurückgreifen, unterschiedliche Typen von Prädispositionen erfassen und damit zu ganz verschiedenen Aussagen führen. Ein Ansatz ist die klassische Kopplungsanalyse, wie sie seit langem mit über das gesamte Genom verstreuten Markern als "genome wide scan" durchgeführt wird und als Ziel die Identifikation eines entsprechenden Gens durch eine Positionsklonierung hat. Der zweite Ansatz prüft in Form einer Fall-Kontroll-Studie, ob bestimmte Allele eines bestimmten Gens zum Auftreten oder zur Manifestation eines Prostata-Karzinoms beitragen und deshalb bei Betroffenen häufiger zu finden sind als bei Kontrollen. Er hat zur Voraussetzung, daß der zu untersuchende DNA-Marker sehr eng benachbart (in der Regel nur einige wenige tausend Basenpaare) bei derjenigen Sequenzveränderung der DNA liegt, die für die Risikoerhöhung verantwortlich ist. Bislang sind solche Assoziationsstudien deshalb nur innerhalb bzw. bei Kandidaten-Genen möglich. Eine genom-weite Suche würde einerseits einen enormen Stichprobenumfang und andererseits eine auf dem heutigen technischen Stand fast unvorstellbare Zahl von Markern (weit mehr als 100 000) erfordern, so daß weitere technologische Entwicklungen abzuwarten sind, bevor ein solcher Ansatz sinnvoll anzugehen ist. Beide Wege wurden in der Abteilung Humangenetik in den letzten Jahren beschritten.
Assoziationsstudien
Da Assoziationsstudien bislang nicht als genomweite Suche durchführbar sind, kann man nur für einzelne Gene prüfen, ob eines der jeweiligen Allele mit erhöhter Suszeptibilität für das Prostatakarzinom einhergeht. Die Auswahl dieser Gene erfolgt sinnvollerweise aufgrund funktioneller Hinweise auf solche Gene, die in Initiation oder Progression des Tumors involviert sein könnten. Für das CAG-Repeat im ersten Exon des Androgen-Rezeptor-Gens ist in der amerikanischen Population eine deutliche, aber nur grenzwertig signifikante Assoziation zwischen Repeatlänge und dem Auftreten eines sporadischen Prostata-CA beschrieben. Da dem Androgen-Rezeptor eine eindeutige Funktion bei der Tumorprogression zukommt, war von Interesse, ob sich diese positive Assoziation auch in einer französisch-deutschen Population bestätigen lässt. Mit einer auf dem ALF-Fragmentanalyser basierenden Untersuchungsmethode analysierten wir 105 Kontrollpersonen, 132 sporadische PCa-Fälle und 28 PCa-Familien (85 betroffene und 46 nicht betroffene Personen), wobei ein dem CAG-Repeat benachbarter GGC-Repeat in die Studie mit einbezogen wurde. Diese Untersuchungen ergaben, dass beide Repeats in der europäischen Bevölkerung eher von untergeordneter Bedeutung sind (Correa-Cerro et al., 1999a).
Auch für einige Polymorphismen im Vitamin D Rezeptor (VDR) Gen wurde in amerikanischen Studien eine positive Assoziation mit PCa nachgewiesen. Wir analysierten im oben benannten Kollektiv drei Polymorphismen und konnten eine schwach signifikante Assoziation zwischen dem heterozygoten Phänotyp eines TaqI-Polymorphismus und PCa in der europäischen Bevölkerung zeigen (Correa-Cerro et al., 1999b).
Veranlaßt durch das hohe relative Risiko für PCa, welches sich aus dem Insulin-like growth factor I (IGF-I) Serumspiegel ermitteln läßt, untersuchten wir weiterhin zwei CA-Repeats im IGF-I-Gen. Allerdings konnten wir keine Assoziation von Allelen des IGF-I Gens mit PCa nachweisen.
Eine Reihe weiterer potentieller Kandidatengene wie z.B. Her2/Neu, PCTA1, RET, MET u.a. wurden und werden in unserer Abteilung unter dem Gesichtspunkt einer erhöhten Suszeptibilität bestimmter Allele für das Prostatakarzinom untersucht.
Kopplungsanalysen
Eine Vielzahl von prädisponierenden familiären Krebsgenen sind in der Vergangenheit über Kopplungsanalysen (Linkageanalysen) entdeckt worden. Bei dieser Art Studien werden polymorphe Mikrosatellitenmarker genutzt um in geeigneten Familien nach Kopplung (gemeinsamer Vererbung von Phänotyp/Krankheit mit Allelen) zwischen ein oder mehreren Markern und einem potentiellen Krankheitslokus zu suchen. In der Regel werden solche Linkagestudien zunächst über das ganze Genom durchgeführt (sog. "genom wide scan"), was einerseits die tatsächliche Existenz eines prädisponierenden Genes beweist und andererseits eine Aussage über die ungefähre chromosomale Lage des Krankheitslokus ermöglicht. Nach Identifizierung einer möglichen Kandidatenregion kann dann durch positionelle Klonierung u.U. auch durch direkte Mutationsanalyse das verantwortliche Gen gesucht und identifiziert werden.
Eine Alternative zur genomweiten Suche bietet die Analyse bereits publizierter Loci am eigenen Familienkollektiv. Diesen Weg verfolgen wir derzeit vornehmlich in unserer Abteilung. Die im Laufe der Zeit von uns gesammelten DNA-Proben von PCa-Familien ermöglichten es uns inzwischen Linkagestudien für die vier wichtigsten prädisponierenden Loci (1p36; 1q24; 1q42 und Xq27-28) durchzuführen.
Die Untersuchung am HPCX-Lokus auf dem X-Chromosom lieferten hierbei die bislang eindeutigsten Resultate. Bei der Untersuchung von 43 Familien mit sechs verschiedenen Markern konnte mit dem DNA-Marker DXS6751 bei einer nonparametrischen Analyse (der Vererbungsmodus wird hierbei vernachlässigt) eine signifikante Kopplung mit einem Z-score von 2,67 bei einer Signifikanz von p = 0,003 zum angesprochenen Locus gezeigt werden (Tabelle 1). Diese Daten deuten demnach auf ein X-chromosomales, prädisponierendes Gen für PCa in dieser Region auf dem X-Chromosom auch in der deutschen Bevölkerung hin. Diese ersten deutlichen Hinweise auf ein für PCa prädisponierendes Gen werden gegenwärtig durch die Analyse weiterer Familien bestätigt.
Tabelle 1: Ergebnisse der Linkageuntersuchungen am HPCX Lokus

Die Daten für die anderen Loci auf Chromosom 1, die durch Untersuchung von jeweils 59 deutschen PCa Familien ermittelt wurden, liefern zum jetzigen Zeitpunkt zwar keine statistisch signifikanten Werte, es zeigte sich allerdings, dass einzelne Familien durchaus auf dem einen oder anderen Lokus koppeln. Dass bei diesen Untersuchungen bei einer relativ bescheidenen Anzahl von Familien keine für sich allein betrachtet signifikante Kopplung über den ganzen Datensatz nachgewiesen werden kann, erscheint bei der enormen Heterogenität des Prostatakarzinoms nicht verwunderlich. Interessanter scheint in diesem Zusammenhang dass im Gegenschluß ein Kopplungsausschluß ebenfalls nicht nachgewiesen werden kann; eine Tatsache, die die Relevanz der untersuchten Genorte auch in der deutschen Bevölkerung verdeutlicht, auch wenn sie vermutlich jeweils nur für einen kleinen Teil der Familien relevant sind.
Funktionelle Analysen
Neben den beiden klassischen Ansätzen Kopplungsanalyse und Assoziationsstudie beschäftigen wir uns seit geraumer Zeit mit der funktionellen Charakterisierung und Untersuchung zweier ausgewählter Kandidatengene PCTA-1 und Caveolin-1.
PCTA-1
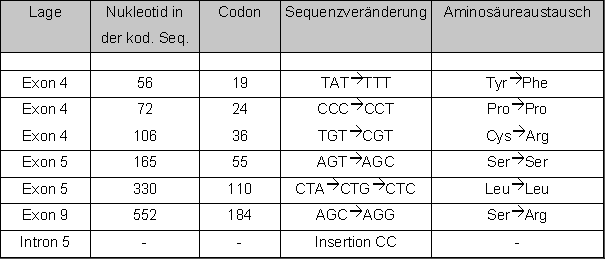
Auf dem PCaP-Lokus (1q42) konnten wir über Datenbankrecherche das Gen "prostate carcinoma tumor antigen 1", PCTA-1 entdecken. PCTA-1 wird selektiv in Prostatatumoren exprimiert (Su et al., PNAS, 93, 7252-7257, 1996), was die Vermutung erlaubt, dass es sich bei PCTA-1 um das gesuchte Kandidatengen für das familiäre PCa in dieser Region handelt. Ausgehend von der veröffentlichten cDNA Sequenz ermittelten wir die komplette genomische Sequenz des über 30kb großen Gens mit 11 Exons (Abb. 1). Nach Charakterisierung der Exon/Intron-Genzen waren wir in der Lage in Familien nach Mutationen in PCTA-1 zu suchen, die aufgrund unserer Linkage-Daten um den Bereich 1q42 auf dem PCaP-Lokus koppeln. Die Mutationssuche wird mit dem "enzymatic mutation detection" EMD-System durchgeführt, welches eigens für diesen Zweck etabliert wurde. Zwar konnten wir bislang noch keine Mutationen nachweisen, fanden aber überraschenderweise eine Vielzahl von exprimierten SNPs ("single nucleotide polymorphisms") (Tabelle 2). Diese SNPs dienen jetzt dazu Assoziationsstudien sowohl in den Familien als auch bei sporadischen Fällen durchzuführen.
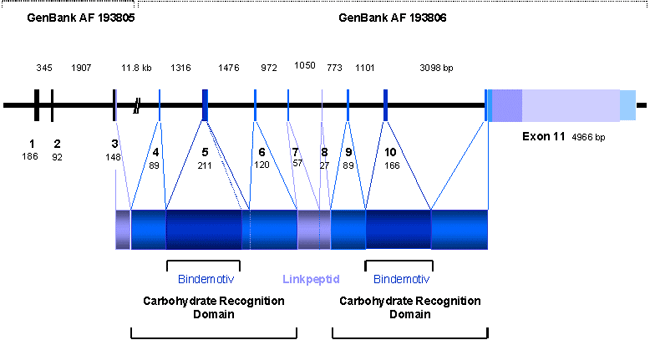
Abb.1 Genomische Struktur von PCTA-1 (nach C.Maier, Abt. Humangenetik). Die Größe der Introns und Exons sowie der Proteinaufbau sind angegeben.
Tabelle 2: SNPs im offenen Leseraster des PCTA-1 Gens
Caveolin-1
Ein weiteres für das PCa relevantes Gen Caveolin-1, wird von uns vornehmlich auf funktioneller Basis untersucht. Methylierungsstudien im Promotorbereich lassen auf eine ausgeprägte Methylierung schließen. Derzeit sind wir dabei diese Daten mit Caveolin-1 Expressionsdaten mehrerer PCa-Zelllinien zu korrelieren. Aufgrund dieser Ergebnisse vermuten wir, dass Caveolin ein neues geprägtes Gen im humanen Genom ist, was aufgrund seiner nachgewiesenen Eigenschaft als Tumorsuppressor weitreichende Konsequenzen in dessen Rolle in der Tumorgenese hätte. Im Rahmen der Expressionsstudien konnten wir zudem alternatives Splicing von Caveolin-1 nachweisen, das offensichtlich zur bereits bekannten ß-Subform des Proteins führt.